ADCs: Function & Pharmacology
This is the second article in a series of five blog posts that are putting ADCs in the spotlight. This article will describe in detail how an ADCs design can control its function and pharmacology. Please read the introductory post ADCs: What are they and why do they matter before this post, if you are unfamiliar with ADCs.
Look out for; ADCs: Pros and Cons, ADCs: Origins and Obstacles, and ADCs: Pipeline and Progress, coming soon.
Key Moieties and their Functions
The mAb, is responsible for the specificity of the ADC, via variable region binding to the target epitope. The mAb moiety retains normal mAb functionality e.g. trastuzumab emtansine has equivalent inherent anti-HER2 effect to trastuzumab [1]. The mAb is also responsible for initial delivery of the payloads into the cell, which occurs by receptor mediated internalisation see Figure 1 below.
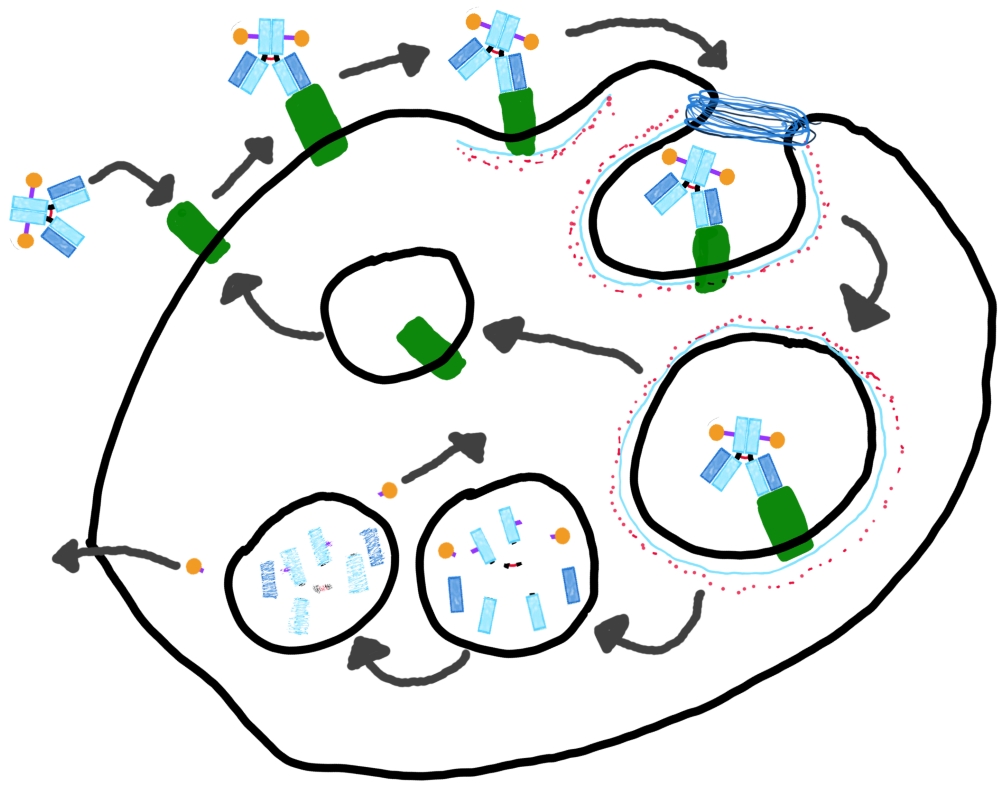
The mAb isotype affects the levels of antibody dependent cell-mediated cytotoxicity (ADCC) and complement dependent cytotoxicity (CDC). IgG1 and IgG2 are ideal isotypes due to increased ADCC and CDC compared with IgG3 and IgG4.
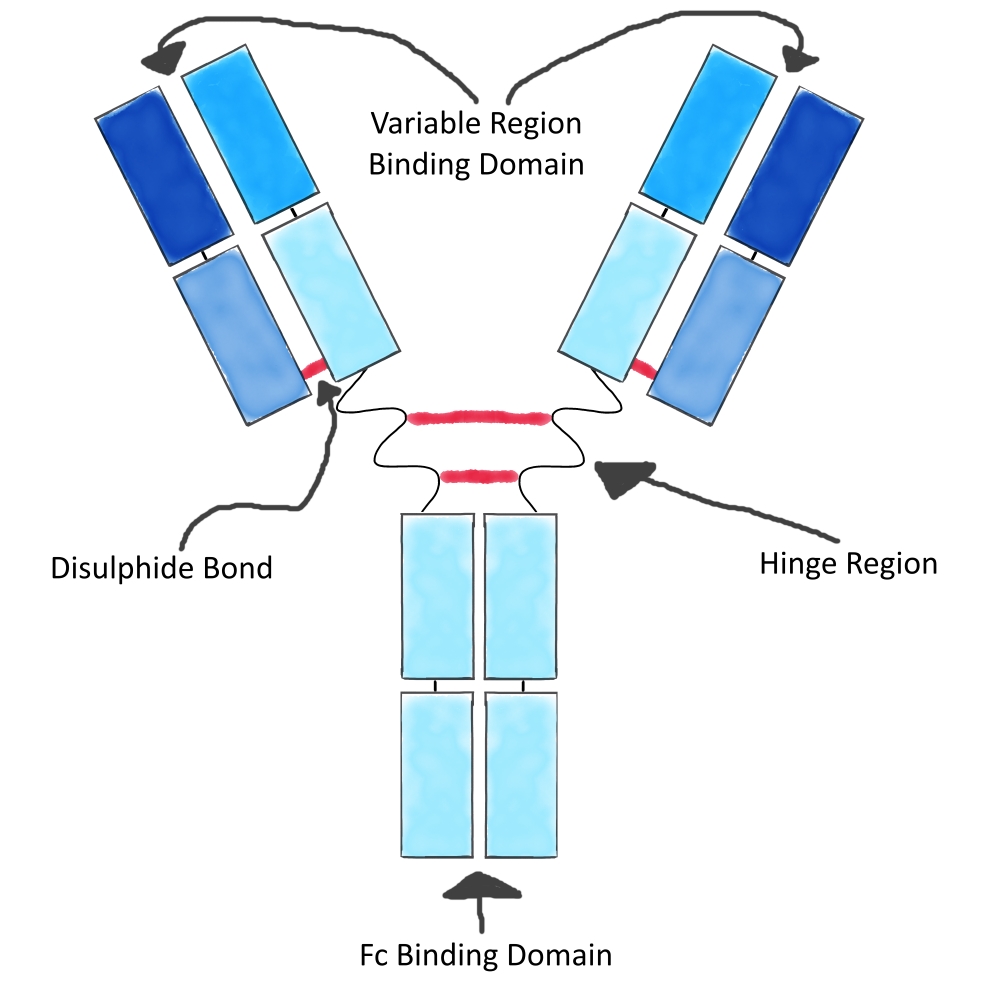
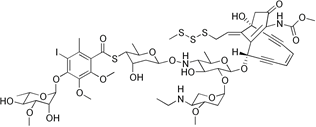
The linkers are responsible for keeping the warheads attached to the mAb and out of systemic circulation, this is especially important as the warheads used in ADCs are too toxic to be administered as unconjugated entities.
Current linker technology attaches to mAbs via lysine or cysteine residues. This results in products with variable DAR. A DAR over 6 is unfavourable due to increased clearance and reduced stability, while a low DAR is unfavourable due to lack of efficacy. Linkers also keep certain payloads inactive whilst bound to the mAb. This is primarily because it prevents the payloads from reaching their own sites of action.
Linkers have at least two sites of conjugation, one to the payload and one to the mAb, some linkers are more complex with cleavage sites within them. The conjugation chemistries used can be the same or different. This level of control in linker design allows warhead release and warhead pharmacokinetics to be fine tuned.

Four typical binding chemistries have been used so far: Disulfide bonds, thioether bonds, dipeptide bonds and pH sensitive bonds. pH sensitive hydrazone bonds are not strongly stable in serum, they are broken at pH of 4.5 or lower.
Disulfide bonds are very stable in serum and release payloads via exchange with other disulphide containing compounds like intracellular glutathione. The rate of exchange is not typically high enough to ensure that desirable warhead concentrations are achieved.
Thioether bonds are also very stable in serum and do not release their payloads at all. Thiother bonded warheads can only be released when the ADC is degraded in the lysosome. The linker along with a lysine residue remains attached to the warhead. The presence of the linker and lysine residue reduces warhead activity and prevents payloads from entering nearby cells.
Dipeptide bonds such as valine-citrulline are stable in serum and are degraded by proteases for example cathepsin B, found in lysosomes. Dipeptide bond cleavage releases the warhead completely, maintaining full activity and ability to enter nearby cells.
Current favoured drug classes for ADCs include maytansinoids, auristatins and calicheamicins.
Trastuzumab emtansine utilises DM-1, a synthetic maytansinoid. Maytansinoids prevent cells progressing through the cell cycle by inhibiting cytoskeleton reorganisation. This is by preventing tubulin dimers polymerising to microtubules.
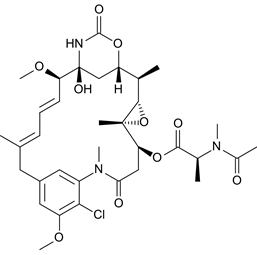
Brentuximab vedotin’s payload is Monomethyl auristatin E, a synthetic auristatin. Auristatins are also antimitotic agents which halt the cell cycle by by preventing tubulin dimers polymerising to microtubules.

Gemtuzumab ozogamicin used calichaemicin, which binds the DNA minor groove to cause double strand scission. This product was removed from the market due to increasing mortality rates compared with alternative therapies.
Payloads differentiate ADCs from mAbs. Warheads can be over 4000 times more potent than conventional chemotherapeutics. Many drug classes previously prohibited for use due to unacceptable toxicity, can now be used as warheads for future ADCs. ADC warheads must be pharmacologically chemotherapeutic. Radioisotopes, cytokines or protein based toxins can be conjugated to mAbs but are not ADCs.
ADCs have potential in many therapeutic fields, due to their potent yet specific pharmacology, and are already making waves in cancer therapy.
References
- Junttila TT, Li G, Parsons K, Phillips GL, Sliwkowski MX. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res Treat. 2011 Jul;128(2):347-56.